脑血管病变
脑血管疾病是危害人类健康的常见病和多发病,且随年龄的增长其发病率和致死率呈上升趋势,因此针对脑血管疾病的防治研究仍然是我国科研工作者和临床医务人员的紧迫任务。
一、胶质细胞和神经元与脑循环调节
人类脑组织代谢活跃,能量消耗大,机体静息状态下消耗总产量20%的能量,并且其耗能量根据机体需求不断变化。在某些疾病状态下,如缺血性脑卒中、蛛网膜下腔出血后血管痉挛、脊髓损伤后继发性缺血等,局部血流减少,葡萄糖或氧供应不足时,会造成神经元和神经胶质细胞损伤甚至死亡。
为了维持神经元的功能,大脑采用了“神经血管偶联”调控机制,即当神经元被激活后,相应区域的血供增加,称为功能性充血。传统理论认为,激活的神经元通过产生某种代谢信号,如氧分压下降、葡萄糖浓度下降或二氧化碳水平升高,使血管扩张,脑血流量增加。最近研究发现,由星形胶质细胞介导的神经递质信号,特别是谷氨酸,在脑血流量调节中具有重要的作用。谷氨酸介导的信号通路促使一氧化氮( nitric oxide, NO )从神经元释放、花生四烯酸( arachidonic acid, AA )衍生物从星状细胞(也有可能是神经元)释放,根据局部氧浓度调节血流量。在大脑的不同区域,谷氨酸介导的信号不同,其作用也各异。
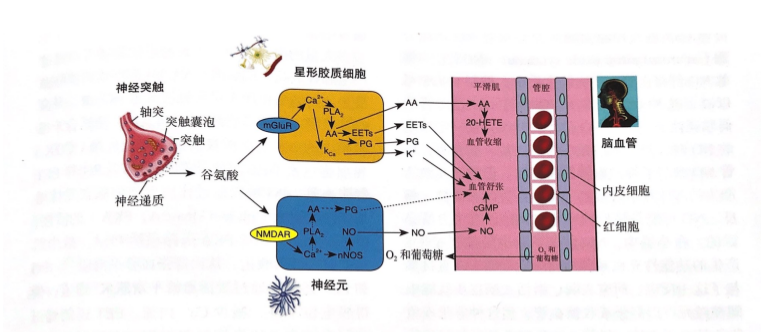
图1 谷氨酸介导的脑血流量调控的主要途径:星形胶质细胞和神经元通过释放神经递质调控动脉对细胞的供氧和葡萄糖
在星形胶质细胞中,谷氨酸通过激活代谢型谷氨酸受体( metabotropic glutamate receptor, mGluR)升高Ca2+浓度,生成AA及其不同的代谢产物:环氧化酶( cyclooxygenase, COX )代谢产生的前列腺素( prostaglandin, PG )和细胞色素P450 ( cytochrome P450,CYP )表氧化酶代谢产生的环氧二十碳三烯酸( epoxyeicosatrienoic acid, EET )舒张血管;ω-羟化酶代谢产生的20-羟二十烷四烯酸[20-hydroxy-(5Z, 8Z, 11Z, 14Z)-eicosatetraenoic acid, 20-HETE] 收缩血管。同时,升高的Ca2+浓度激活Ca2+依赖性钾通道“Ca2+ dependent K+ channel, KCa”, 释放K+ ,舒张血管。在神经元中,突触释放谷氨酸作用于N-甲基-D-天门冬氨酸受体(N methy-D aspartate receptor, NMDAR)使Ca2+浓度升高,促使神经元型一氧化氮合酶(neuronal nitric oxide synthase, nNOS )释放NO,激活平滑肌鸟苷酸环化酶,生成环磷酸鸟背(Cycic guanosine monophosphate, cGMP)舒张血管。Ca2+浓度的增高也能使磷脂酶A2 ( popolipase A2, PLA2)的代谢产物AA增多,经COX2及表氧化酶代谢分别生成PG和EET舒张血管。
二、内源性小分子物质与脑循环调节
1980年,研究者们发现只有当血管内皮细胞层完整时,乙酰胆碱( actylcholine Ach)才能引起血管舒张;当内皮被剥离后,Ach导致血管收缩。因此,推测Ach刺激内皮细胞产生“内皮来源的舒张因子(endothelium -derivedrelaxing factor, EDRF )”作用于血管平滑肌细胞( vascular smooth muscle cell, VSMC ),使其舒张,扩张血管。
1987年,Palmer首次发现了最著名的EDRF-NO。 随后陆续有了其他EDRF相关的研究报道,包括一氧化碳(CO)、硫化氢(H
2S)、过氧化氢(H
2O
2)、K
+等。
三、
钾离子通道与脑循环调节
K
+通道的活性在血管张力调控中起着重要的作用。血管平滑肌细胞的K
+通道激活导致细胞膜超极化,电压门控Ca
2+通道关闭, 细胞内Ca
2+浓度下降,血管舒张。体外检测脑血管平滑肌膜电位为-40~-70mV,但缺乏相应的体内检测数据。仅仅几毫伏的膜电位变化可以使血管张力产生巨大的变化。被激活的K
+通道能介导脑血管对不同刺激因素产生不同的效应,包括受体结合反应、第二信使信号通路和缺氧反应等。内皮依赖性超极化因子( endothelium-dependent hyperpolarizing factor, EDHF )通过激活K
+通道使血管平滑肌超极化,舒张血管。
大量的研究表明,在大脑中,不仅血管平滑肌表达多种K
+通道,内皮细胞也表达K
+通道。内皮细胞超极化,调节Ca
2+浓度,是介导EDHF释放的重要机制之一。
四、
脑血管在不同状态下的病理生理变化
血管内皮细胞对于循环系统稳态和各种血管功能有十分重要的调节作用,特别是在大脑中。关于内皮功能障碍和血管疾病的机制研究不断取得新的认识。RAAS系统激活、氧化应激和局部炎症是致病的关键分子机制。内源性的血管保护机制则包括抗氧化分子、抗炎分子和过氧化物酶体增殖物激活受体( peroxisome proliferator-activated receptor, PPAR )。尽管已经发现了它们的重要性,但是这些分子在脑血管疾病中的具体机制研究远落后于整体血管生物学研究。进一步阐明内源性分子和信号通路的血管保护机制,将为脑血管病变的靶向预防和治疗,特别是脑卒中和老年痴呆症防治研究提供基础。
(一)
高血压
1.
急性高血压
急性高血压多发生于高血压急性发作期、嗜铬细胞瘤、升压药导致的全身性血压增高或头部损伤。急性高血压不仅会损害内皮细胞的形态,还会削弱脑血管对乙酰胆碱诱导的内皮依赖的血管舒张反应,而对硝普钠诱导的血管舒张反应影响较小。急性高血压和脑外伤时大脑中超氧阴离子的产生增加。局部给予SOD可以使脑血管对乙酰胆碱诱导的内皮依赖性血管舒张反应恢复至正常,提示超氧阴离子使NO失活,导致内皮功能损伤。
2.
慢性高血压
最近一项遗传学分析表明,受损的内皮依赖性舒张反应与卒中易感表型具有相关性,提示正常的内皮功能在脑血管中起着保护作用。慢性高血压患者和模型动物的外周血管的内皮依赖性舒张反应均受到损伤。与内皮依赖性反应相比,慢性高血压状态下脑血管对硝酸甘油、硝普钠和腺苷等的非内皮依赖性反应并未受损。这表明脑血管功能障碍发生在内皮细胞,而不是平滑肌细胞。
研究者们围绕慢性高血压脑血管内皮依赖性舒张反应损伤的机制展开了一系列研究。降血压治疗可以恢复内皮依赖性反应至正常水平,提示这种损伤是可逆的。慢性高血压脑动脉中也存在类似的病理生理机制。
(二)
高胆固醇血症和动脉粥样硬化
高胆固醇血症和动脉粥样硬化患者和模型动物的非脑动脉血管的内皮依赖性舒张功能受损。无论是在基础状态下,还是在内皮依赖性激动剂刺激下,EDRF的产量或活性都受到了损害。一些研究发现,即使仅是在尚未发生动脉粥样硬化病变的单纯高胆固醇血症的情况下,也足以使得内皮细胞功能障碍。
动脉粥样硬化对脑血管内皮依赖性反应的影响尚不明确。据观察,动脉粥样硬化的颈动脉内皮依赖舒张反应性下降,当动脉粥样硬化好转时,颈动脉的内皮依赖舒张反应性也随之恢复。在动脉粥样硬化动物模型中,尽管颈动脉和主动脉内皮依赖性舒张反应受损明显,但相同动物的脑血管内皮依赖性舒张反应可以维持在正常水平。
体内研究表明,动脉粥样硬化也可以使脑部大血管和眼部供血血管对活化的白细胞收缩反应增强。活化的白细胞可释放多种血管活性物质(包括ROS、血栓素、白三烯、血小板活化因子和内皮素),导致动脉粥样硬化的过程中脑血管收缩反应增强。
(三)
糖尿病
动物实验和人体研究发现,糖尿病时不仅颅外血管的内皮功能出现异常, 脑动脉和基底动脉的舒张功能也发生异常。糖尿病血管功能损伤的机制和全身性血管功能损伤的机制不同。糖尿病状态下脑血管对内皮非依赖性刺激因子(如硝酸甘油、硝普钠和毛喉素)的血管舒张反应是正常的,提示不存在血管非特异性损伤。相比之下,糖尿病状态下,脑血管对某些直接作用于血管平滑肌的刺激因子(包括异丙肾上腺素、去甲肾上腺素及ATP敏感钾通道激活剂)的舒张反应受损。
(四)
缺血
缺血可以损害脑血管对不同刺激因子的反应。脑动脉缺血后再灌注会导致对乙酰胆碱的内皮依赖性血管舒张反应受损。SOD和过氧化氢酶可以改善受损的脑血管内皮依赖性血管舒张反应,提示超氧阴离子等ROS的产生是缺血后再灌注内皮功能损伤的机制之一。
(摘自《血管生物学》第2版)